Introduction
SIB-BLAST is a novel algorithm developed to overcome the model corruption problem that occurs frequently in the later iterations of PSI-BLAST searches.The algorithm compares resultant hits from iteration two and the final iteration of a PSI-BLAST search, calculates the figure of merit for each "overlapped" hit and re-ranks the hits according to their figure of merit. The premise of the algorithm is based on the observation that the profile, namely, the position specific scoring matrix (PSSM), in the first two rounds of a PSI-BLAST search, is the least corrupted since it is comprised mostly of close homologs. These profiles are used to search for more distant homologs, which are used to generate subsequent PSSMs. As more distant homologs are incorporated into the PSSM, non-homologous sequences frequently get included also, thus leading to model corruption. Hence, "benchmarking" hits from later iteration against earlier round when the model is least corrupted should improve the accuracy of a PSI-BLAST search.
Method
SIB-BLAST is comprised of the following steps:
- performing a PSI-BLAST search of a query protein sequence against the non-redundant database;
Note: algorithm parameters used for PSI-BLAST searches are set to default values except for the following: - Expect threshold is preset to 1000 instead of the default value 10.
- Number of target sequences reported is user-defined and is restricted to a maximum of 20000.
- comparing resultant hits in iteration 2 and the last iteration;
- calculating a figure of merit for each hit by combining its corresponding E-values at iteration 2 and at the final round;
Please see below for an explanation of the figure of merit.
- re-ranking the merged list of hits according to their figure of merit.
- Expect threshold is preset to 1000 instead of the default value 10.
Input
SIB-BLAST requires three inputs from the user:
- INPUT SEQUENCE : either enter a single protein sequence in FASTA format into the textbox or upload it in a file.
- ITERATIONS : the number of iterations of PSI-BLAST search to be performed.
- MAXIMUM HITS : maximum number of target sequences to be reported. This number is purposefully restricted to be higher than the PSI-BLAST default value of 500 to ensure that even weak hits are reported in the individual rounds that might become significant once results from different rounds are combined.
A sequence in FASTA format contains a single-line description with its FIRST character being a greater-than (">") symbol. Sequence data starts at the next line, with each line preferably containing no more than 80 letters.
Example of a protein sequence in FASTA format:
>gi|12345|TEST sequence
MAKNNAVAGFNALNGVELNLFTTDELKAIHYATMEVLMDPGIQVSDPEARQIFKENGCEVNEKTNVVKIP
EYLVRKALQLAPSRFVLWGRDKKFNTVQECGGKVHWTCFGTGVKVCKYQDGKYVTVDSVEKDIADIAKLC
DWAENIDYFSLPVSARDIAGQGAQDVHETLTPLANTAKHFHHIDPVGENVEYYRDIVKAYYGGDEEEARK
KPIFSMLLCPTSPLELSVNACQVIIKGARFGIPVNVLSMAMSGGSSPVYLAGTLVTHNAEVLSGIVLAQL
TVPGAKVWYGSSTTTFDLKKGTAPVGSPELGLISAAVAKLAQFYGLPSYVAGSOSDAKVPDDQAGHEKTM
TTLLPALAGANTIYGAGMLELGMTFSMEQLVIDNDIFSMVKKAMQGIPVSEETLAVESIQKVGIGNNFLA
LKQTRQLVDYPSNPMLLDRHMFGDWAAAGSKDLATVAHEKVEDVLKNHQVTPIDADIFKDMQAIVDKADK
AFRGM
NOTE: sequence alphabets can be in either upper/lower case
Please choose from the drop-down box any number between 3-10.
NOTE: As noted by the PSI-BLAST developers at NCBI, it is recommended to run no more than five to six iterations of PSI-BLAST (Schaffer et al., 2001).
Please choose from the drop-down box for a choice of 1000, 2000, 5000, 10000, or 20000 sequences.
Output
SIB-BLAST output is a rank-ordered list of putative homologs of the query sequence found in the non-redundant protein database. For the test sequence described above it is:
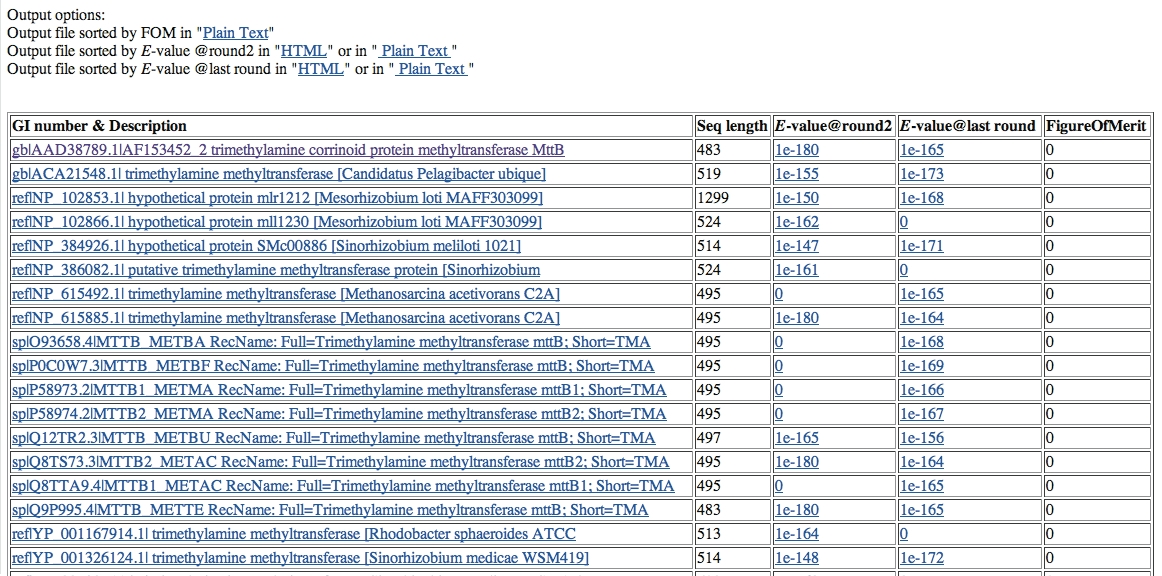
Explanation of output parameters:
GI number: this is the sequence identification number assigned to each sequence record processed by NCBI.
E-value at round 2: this is the E-value (Expectation value) reported by PSI-BLAST for round 2 of the search.
E-value at last round: this is the E-value reported by PSI-BLAST for the last iteration (defined by user).
Figure of Merit: this is the "combined" E-value of iteration 2 and the last iteration.
Based on empirical analysis of the Aravind model test set (Aravind and Koonin, 1999), an empirical threshold at which errors appeared at an accelerated rate is at a FOM of ~ 10-8. However, it should be noted that this FOM threshold is expected to be dependent on the database size.